Die Bewertung des Nutzen-Risiko-Verhältnisses eines Arzneimittels stellt ein zentrales Element bei der Begutachtung im Rahmen der Arzneimittelzulassung dar. Besonders bei Impfungen spielt dies eine zentrale Rolle, wie am Beispiel von Celvapan aufgezeigt wird.
Von Barbara Tucek und Petra Falb*
Das sogenannte Benefit-risk assessment, die Bewertung des Nutzen-Risiko-Verhältnisses eines Arzneimittels, stellt ein zentrales Element in der Begutachtung der Arzneimittel-Zulassung dar; dies wird jedoch nicht nur vor der Zulassung, sondern auch regelmäßig danach – sowohl anlassbezogen als auch routinemäßig – durchgeführt. Nur wenn der Nutzen das potenzielle Risiko eines Arzneimittels überwiegt, erfolgt die Zulassung beziehungsweise bleibt sie aufrecht.
Um heutzutage ein Zulassungsverfahren für einen Impfstoff erfolgreich abschließen zu können, sind von Seiten der Antragsteller umfassende gesetzliche und wissenschaftliche Anforderungen zu erfüllen. Für jede Arzneispezialität ist ein Dossier vorzulegen, das sich aus folgenden Bausteinen zusammensetzt: ein regulatorischer Abschnitt, der aus administrativen Daten, Fach- und Gebrauchsinformation sowie der Kennzeichnung besteht und ein Qualitätsteil mit den Daten zur Herstellung sowie präklinische und klinische Daten. Als Grundlage für die Bewertung durch die Behörde dienen relevante europäische und österreichische Gesetzestexte, regulatorische und wissenschaftliche Guidelines der Europäischen Arzneimittelagentur (European Medicines Agency, EMA), das Europäische Arzneibuch, Leitlinien und Empfehlungen der WHO sowie die aktuelle wissenschaftliche Fachliteratur.
Bei zugelassenen Arzneimitteln sind in der jeweiligen Fachinformation („Austria-Codex“ oder im Arzneispezialitätenregister des Bundesamtes für Sicherheit im Gesundheitswesen (BASG)/AGES Pharm-Med http://pharmaweb.ages.at/index.jsf) alle Nebenwirkungen aus den klinischen Studien, den Sicherheitsstudien nach Zulassung und aus Spontanberichten, zu welchen nach Bewertung durch die Behörde ein möglicher kausaler Zusammenhang hergestellt werden kann, gelistet. Dies wird regelmäßig überprüft und, wenn notwendig, überarbeitet. Die Informationsgrundlage für Impfstoffe ist für Ärzte, Apotheker und medizinisches Fachpersonal – wie bei anderen Arzneimitteln auch – die Fachinformation. Diese enthält ausführliche fachlich relevante Passagen, die im Gegensatz dazu in der Gebrauchsinformation („Beipackzettel“), die für den Patienten bestimmt ist, aus Lesbarkeits- und Verständlichkeitsgründen bewusst nicht immer enthalten sind.
Sicherheitsprofil von Impfstoffen
Neben präklinischen Studien, in denen erste Erkenntnisse am Tiermodell gewonnen werden, sind für die Zulassung auch klinische Studien gemäß den gesetzlichen und wissenschaftlichen Anforderungen durchzuführen. Hierbei variiert die Probandenzahl zwischen 1.000 und 75.000; je nachdem, ob es sich um einen bekannten (wie zum Beispiel Tetanus, Diphtherie, FSME) oder um einen hochinnovativen, neuen Impfstoff (beispielsweise HPV, Rotaviren) handelt. Jedoch werden sehr seltene Nebenwirkungen in diesem Rahmen statistisch nicht bzw. nur unzureichend erfasst. Grundsätzlich gilt folgende Faustregel: Um eine Nebenwirkung bei einer von 1.000 Versuchspersonen zu erfassen, benötigt man 3.000 Versuchspersonen, um diese Aussage mit 95-prozentiger Sicherheit tätigen zu können.
Unter „Pharmakovigilanz“ versteht man die nach der Zulassung einsetzende Überwachung von Arzneimitteln am Markt, das heißt das Monitieren des Nutzen-Risiko-Profils. Dies geschieht mittels Registrierung und Bewertung von Spontanmeldungen, angeordneter Sicherheitsstudien, Signaldetektion, weltweiter Datenbankrecherche, Beurteilung der von den Zulassungsinhabern regelmäßig vorgelegten Periodic Safety Update Reports (PSUR), einer erneuten Nutzen-Risiko-Bewertung binnen einer gesetzlichen Frist nach Zulassung/Vermarktung und einer engen Kooperation mit den EU-Mitgliedsstaaten. Zunehmend werden Pharmakovigilanzmaßnahmen wie zum Beispiel Sicherheitsstudien im Verlauf des Zulassungsprozesses im Rahmen des Risk Management Plans mit dem künftigen Zulassungsinhaber vereinbart (www.ema.europa.eu/pdfs/human/euleg/9626805en.pdf). Dies erfolgte auch für die Zulassung von Celvapan (Influenza A(H1N1) Impfstoff), wo als Teil dieses Risk Management Plans eine prospektive Kohortenstudie an zumindest 9.000 Patienten in unterschiedlichen Altersgruppen, einschließlich Patienten mit geschwächter Immunabwehr, als erforderlich erachtet wurde, um das Wissen über die Sicherheit dieses neuen Impfstoffes weiter vertiefen zu können. Unbestritten ist, dass meist erst durch die breite Anwendung in der Bevölkerung sehr seltene Nebenwirkungen erfasst werden können.
Ebenso ist es bei der Sicherheitsbewertung nach Markteinführung unerlässlich, die Hintergrund-Inzidenzraten bestimmter Erkrankungen je nach Land, Alter und Geschlecht zu kennen, um eventuelle impfstoffbedingte Nebenwirkungen davon abgrenzen zu können.
Die Publikation „Importance of background rates of disease in assessment of vaccine safety during mass immunisation with pandemic H1N1 influenca vaccines“ (Lancet, 31 Oct 2009) berichtet über die Bedeutung der Hintergrund-Inzidenzraten und deren Interpretation hinsichtlich Impfstoffsicherheit am Beispiel einer Massenimmunisierung mit pandemischem H1N1-Influenza-Impfstoff (siehe Tab. 1). Es ist bekannt, dass rund zwölf Prozent der Schwangerschaften in Spontanaborten münden. Dies bedeutet, dass pro einer Million geimpfter Schwangerer mit 397 Spontanaborten binnen eines Tages nach der Impfung beziehungsweise 16.684 Spontanaborten binnen sechs Wochen nach Impfung zu rechnen ist, unabhängig von dieser H1N1-Impfung.
Speziell bei Impfstoffen ist zu beachten, dass hauptsächlich gesunde Personen, besonders Kinder, geimpft werden, die eine hochsensible Population darstellen. Die Nutzen-Risiko-Analyse wird von den Zulassungsbehörden für die Gesamtzahl der zu behandelnden Personen durchgeführt, wohingegen der individuelle Nutzen für den einzelnen Patienten vom Arzt bewertet werden muss.
Generell nimmt die Anzahl der Nebenwirkungs-Meldungen zu. Dies ist unter anderem darauf zurückzuführen, dass heute einerseits wesentlich mehr Impfstoffe zugelassen sind als in den letzten Jahrzehnten und somit mehr Dosen verimpft werden, andererseits ist jedoch in der Bevölkerung sowohl ein Anstieg der Aufmerksamkeit gegenüber Impfreaktionen als auch eine Verringerung ihrer Akzeptanz (zum Beispiel bei Lokalreaktionen) zu verzeichnen. Außerdem wächst das Bewusstsein zur Meldepflicht bei Angehörigen der Gesundheitsberufe.
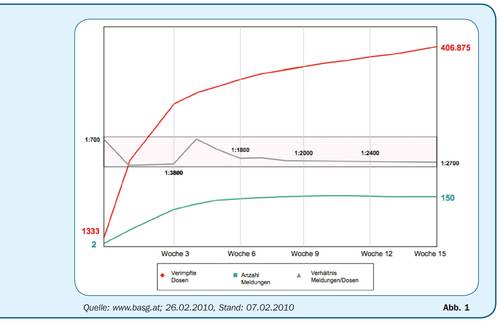
Den Zusammenhang zwischen gemeldeten Nebenwirkungen zu verimpften Dosen anhand des Pandemie-Impfstoffes Celvapan zeigt Abb. 1. Dieser Grafik ist zu entnehmen, dass, obwohl die Anzahl an Nebenwirkungen über 15 Wochen von zwei auf 150 ansteigt, das Verhältnis gemeldeter Nebenwirkungen zu verimpften Dosen trotzdem relativ konstant bleibt (1:700 bis 1:2.700), da sich gleichzeitig die Anzahl der verimpften Dosen von 1.333 auf 406.875 erhöht. Weiters ist eine höhere Anzahl an Spontanmeldungen innerhalb der ersten Wochen nach Beginn der Impfkampagne zu erkennen. Dies ist in erster Linie auf die verstärkte Aufmerksamkeit seitens der Ärzte und Patienten zurückzuführen – ein bekanntes Phänomen nach Einführung eines gänzlich neuen Impfstoffes.
Wird eine unerwünschte Arzneimittelwirkung (UAW) gemeldet, bei der nach Begutachtung durch die Behörde ein Kausalzusammenhang nachgewiesen oder zumindest nicht ausgeschlossen werden kann, sind folgende Maßnahmen möglich:
- Eine Information der Anwender (Ärzte, Apotheker, medizinisches Fachpersonal)
- Aufforderung zur Stellungnahme des Zulassungsinhabers
- Ein Update der Fach- und Gebrauchsinformation
- Involvierung der Mitgliedsstaaten bei EU-weiten Zulassungen
- Einschalten der Pharmacovigilance Working Party/EMA
- Auflagen für Zulassungsinhaber sowie im Extremfall die
- Aussetzung oder Rücknahme der Zulassung
Derzeit sind in Österreich rund 90 Impfstoffe zugelassen, die alle das Prädikat „hohe Sicherheit“ verdienen. Diese Impfstoffe haben nicht nur ihre Wirksamkeit, sondern auch ihre Sicherheit zu beweisen, und das nicht nur vor, sondern auch nach der Zulassung.
Das wichtigste Instrument zum zeitnahen Erkennen von Risikosignalen ist die passive Überwachung, das heißt die konsequente und gewissenhafte Meldung von unerwünschten Arzneimittelwirkungen. Gemäß dem Arzneimittelgesetz und der Pharmakovigilanz-Verordnung sind vermutete schwerwiegende Nebenwirkungen, das heißt:
• tödlich
• lebensbedrohend
• stationäre Behandlung oder deren Verlängerung erforderlich
• bleibende oder schwerwiegende Behinderung oder Invalidität
• kongenitale Anomalie oder Geburtsfehler
unverzüglich zu melden. Dies betrifft auch Reaktionen, die in Häufigkeit und Intensität das bekannte Nebenwirkungsprofil überschreiten. Die entsprechenden Meldeformulare findet man auf der Website des BASG/AGES PharmMed www.basg.at; dort sind auch allgemeine und aktuelle Informationen zu Impfstoffen veröffentlicht.
|
Tab.1: Vorhersage koinzidierender, zeitlich assoziierter Events nach Einzeldosis einer hypothetisch verabreichten Vaccine |
|||
|
Anzahl koinzidierender Events nach Impfdosis innerhalb von: |
Guillain-Barré Syndrom je 10 Mio Geimpfter |
Spontanaborte je 1 Mio geimpfter Schwangerer |
Opticusneuritis je 10 Mio geimpfter Frauen |
|
1 Tag |
0,51 |
397 |
2,05 |
|
7 Tagen |
3,58 |
2.780 |
14,40 |
|
6 Wochen |
21,50 |
16.684 |
86,30 |
|
Zugrundeliegende baseline Rate |
1,87 pro 100.000 Personen-Jahre, alle Altersgruppen (UK) |
12 % der Schwangerschaften |
7,5 pro 100.000 Personen-Jahre, Frauen (US) |
|
*basierend auf Hintergrund-Inzidenzraten |
|||
Quelle: „Importance of background rates of disease in assessment of vaccine safety during mass immunisation with pandemic H1N1 influenca vaccines“ St. Black, J. Eskola, C. Siegrist et al; Lancet, 31 Oct 2009
|
„Wenn behauptet wird, dass eine Substanz keine Nebenwirkung zeigt, so besteht oftmals der Verdacht, dass sie auch keine Hauptwirkung hat.“ |
*) Dr. Barbara Tucek, Mag. Petra Falb:
beide: AGES PharmMed, Institut Zulassung & Lifecycle Management,
Schnirchgasse 9, 1030 Wien;
Tel. 050/555-36 540;
E-Mail: barbara.tucek@ages.at, petra.falb@ages.at